
Evidence-based maintenance of medical devices: Current shortage and pathway towards solution
1.Introduction
Today’s rapid digital transition and technological evolution affects various sectors including healthcare, causing the advancement in medicine [1]. Technological evolution in healthcare enables more precise early diagnosis resulting in personalized and effective patient treatment. The term medical technologies cover services or solutions, devices and equipment that vary from high-risk to low-risk category developed to solve a health problem and improve quality of life. The use of increasingly sophisticated medical technology in healthcare comes with the need for clinical engineers – “professionals who support and advance patient care by applying engineering and managerial skills to healthcare technology” [2]. In order to ensure the continued safe-use of a device for the manufacturer’s intended purpose these professionals must know how to use it, have means to identify when it is not used correctly or when it no longer works at manufacturer’s defined parameters.
Along with the digital transition, the medical devices industry has grown rapidly and incessantly over the past century. According to the World Health Organisation (WHO), today, there are an estimated 2 million different kinds of medical devices (MDs) on the world market, categorized into more than 7000 generic devices groups [3]. The complexity of the medical technology assets found in healthcare, in terms of number and diversity, is reflected in the complexity of technology management, which must be efficient so that the technology can always be used according to its intended use, safely and appropriately. However, for the health industry, be it hospitals or manufacturers of medical devices, nothing counts more than the safety of a patient.
2.Medical device lifecycle: Pre- and post-market
In countries around the world, MDs undergo a distinctive process from ideation, design and development phase, up to testing, approval and certification before the production, production process itself and post-market surveillance (PMS). Produced medical devices must comply with health, safety and environmental protection requirements defined in regional and/or national directives and regulations.
Manufacturers of the medical devices are legally responsible that the requirements of the directives and regulations for every phase of medical device life cycle are met. To ensure legal requirements are met and high-quality products are produced, manufacturers are required to implement quality management systems defined per international standards such as ISO 13485 – Medical devices – Quality management systems – Requirements for regulatory purposes, ISO 14971 Medical devices – Application of risk management to medical devices, etc. Additionally, a set of safety and technical standards referring to specific medical device requirements have been developed such as IEC 60601 Medical electrical equipment, ISO 15223 Medical devices – Symbols to be used with information to be supplied by the manufacturer, ISO 10993 Biological evaluation of medical devices, IEC 62304 Medical device software – Software life cycle processes, IEEE Standard for Health Informatics – Point-of-care medical device communication - Part 10201, etc. [4].
Administrative marking has been introduced to indicate that medical devices conform with health, safety, and environmental protection requirements. The administrative marking is introduced differently from country to country. For example, in China, CCC (China Compulsory Certification) [5] marking is used for labelling of approved devices. Japan uses PSE [6] (Product Safety Electrical Appliance and Material) marking. In Australia [7], there are various types of CE markings, or their equivalents used. In the EU CE mark is introduced and serves as an administrative marking that indicates conformity with health, safety, and environmental protection standards for products sold within the European Economic Area (EEA). Since leaving the EU, the United Kingdom (UK) [8] has introduced the new mark UKCA (United Kingdom Conformity Assessed) which is enforced from 2021. Canada enforces its own CSA [9] (Canadian Standards Association) marking. In the United States, equivalent to Europe’s CE [10] marking is UL (Underwriters Laboratories) marking. Generally, the administrative marking is issued by authorized notified bodies. In all regions, the manufacturer governs the overall process from ideation to market, whereas independent authorized third parties check for compliance, give approval for commercialization and certification before placing the device on the market. In this way, the process of approval and certification of the product’s compliance with directives and regulations during pre-market process is independent and standardized ensuring public guarantee from the point of view of safety and accuracy of the use of medical devices. So, it is evident that MDs are produced in a strict process harmonized with international standards, and put on the market after authorization and marking.
Specifically, in the European Union (EU), manufacturers of the medical devices are legally responsible that the requirements of the Medical Device Directives (MDD) and Medical Device Regulations (MDR) are met. The directives, in conjunction with the harmonized standards, form a framework for manufacturers to develop a comprehensive feedback system intended to ensure the continued safe-use of a device for the manufacturer’s intended purpose. The certification process confirms conformity of the manufacturer and the device itself to these requirements. It is issued by the European Notified Bodies [11], organizations appointed by EU countries to assess the compliance of products before they are placed on the market. The CE mark is an indication that the product is in conformity with the directive and regulation. It can be effortlessly identified by authorities and customers using these means and having it well visible on the product. Furthermore, the medical device may not be placed on the market in the EU and EEA without the CE marking.
As mentioned, during the pre-market phase, MD functionality, safety and performance needs to be designed to satisfy multiple requirements defined in a variety of international standards such as basic safety standards, electro compatibility standards, biocompatibility standards, environmental protection standards, etc. Pre-market evaluation and classification of product quality, safety and performance is conducted by manufacturers and/or authorized, notified bodies. For the EU market, the device entering the market has clear classification into risk category and compliance marking such represented as CE mark.
Device risk assessment during pre-market is done by manufacturers based on pre-market data including: design, clinical trials and test-model assumptions which may not accurately represent real-life situations. All of the activities of pre-market testing are conducted in a controlled environment where the device is completely isolated from external influences such as usage, changes to the usage methods and patterns, humidity and temperature changes and influences of electromagnetic fields of surrounding devices. In addition, pre-market data of device assessment typically reflects ‘short term’ periods of observation or use, and may not reflect potential incidents that would arise over longer periods of usage. Although medical devices are designed, developed, manufactured and distributed on the global market after thorough pre-market evaluation, residual risks regarding safety and performance will remain throughout the product’s lifetime. This is due to a combination of factors, such as inherent product variability, factors affecting the medical device’s use, environment, different user interaction, as well as, unforeseen medical device failure or misuse. Design and development activities for medical devices ensure the residual risks are acceptable with respect to anticipated benefits before the product is released on to the market. This limitation is causing unfortunate incidents and injuries involving medical devices once they are being used. As medical devices and human–system interactions become more complex, usability issues are a persistent challenge and are gaining more recognition for their impact. A 2016 Johns Hopkins study reported that medical errors are the third leading cause of death in the United States [12]. Prioritizing human factors is imperative to decrease the occurrence of design flaws, eliminate or reduce use-related hazards, improve patient adherence and ultimately ensure safe outcomes for end users of medical devices.
To facilitate prevention of these adverse events involving medical devices regulators prescribed a post-market surveillance (PMS) [13]. PMS is defined as a set of activities to collect and evaluate experience gained from medical devices that have been placed on the market, and to identify the need to take any action. It can be seen as a collection of processes and activities used to monitor the performance of a medical device once it is placed in healthcare institutions. These activities are designed to generate information regarding the utilization of the device, to expediently identify device design and/or usage problems and accurately characterize the device behaviour in practice. Vigilance, management and maintenance of medical devices can be seen as set of activities within the PMS mechanism currently being implemented globally. The vigilance system involves cooperation of different stakeholders: users, manufacturers, competent authorities, and others that can effectively facilitate the detection of previously unknown adverse product information and prevent future recurrence of incidents that might have otherwise led to death or serious deterioration in health. Another aspect of PMS is seen in medical device management and maintenance in healthcare institutions, again done by manufacturers/distributors and healthcare institutions itself.
The WHO Global Model Regulatory Framework for Medical Devices [3], including in vitro diagnostic medical devices, like many other international regulatory frameworks, requires the implementation of PMS systems. It requires that receiving and evaluating feedback are the minimum requirements for post-market surveillance, but that this can be expanded to include other activities. The WHO Global Model Regulatory Framework for Medical Devices also includes the activities of the national regulatory authorities (NRAs) acting in response to reports of adverse events received, so-called vigilance. Thus, the terms post-market surveillance, vigilance and market surveillance are closely linked.
Given the abovementioned limitations in pre-market device risk classification, rising challenges in healthcare sector seen through pandemics and adoption of sophisticated technology in healthcare, regulatory bodies worldwide are increasingly emphasizing that PMS data should be used for usability-related improvements during the product development process. It remains important to continue to collect and evaluate information on the medical device during production and post-production to meet requirements for the monitoring of products and processes and to ensure the residual risks remain acceptable with respect to benefits. However, even though this requirement is sound, a technical obstacle is preventing approach to take on full impact. The reason for this is that PMS differs significantly worldwide. While pre-market processes are harmonized and standardized globally, the challenge of medical device conformity assessment in PMS is becoming more emphasized due to the fact that it has a direct impact on diagnostics and treatment of patients. How so? It is seen that medical devices are produced following harmonized, standardized and regulated premarket processes. Also, PMS is conducted by manufacturers or distributors as defined within directives and regulations. However, it is not unlikely that an error in MD performance causes patient injury or in the worst-case scenario patient death. So, if there is no difference on medical devices between countries why does their performance differs which is noticeable by the number of incidents reported?! If medical devices are the same – produced and assessed according to directives/regulation, monitored by PMS – the only reason for the situation can be found in different approaches to medical device management, including preventive service. This indicates that current PMS processes possibly have gaps to be addressed. Getting the right balance between premarket and post-market data collection – specifically, where appropriate, a greater reliance on post-market collection, including real-world data collection, can reduce the extent of premarket data collection and directly impact when patients will have access to high-quality, safe and effective medical devices. But, greater reliance on post-market data collection could undermine patient safety if the necessary and timely data collection does not occur.
In the EU, the new Medical Device Regulation [14, 15, 16] regulates PMS more strategically with the intention to address these challenges. MDR introduces independent, third party – notified body [11] and prescribes that some of PMS activities should be performed by these bodies. In respect to the new MDR, the EU Commission urged member states to tighten controls, increase surveillance, and restore full confidence in the EU CE marking regulatory system. The commission proposed the following:
• Verify that NBs are designated only for the assessment of medical devices and technologies that correspond to their proven expertise and competence.
• Ensure that all NBs exercise the authority given to them by law to ensure that manufacturers conform to regulations through assessment (e.g. power to conduct unannounced inspections).
• Reinforce market surveillance by national authorities (e.g. spot checks for certain types of devices).
• Improve the impact of the vigilance system for medical devices:
– Provide systematic access for NBs to reports of adverse events.
– Encourage healthcare professionals and patients to report adverse events.
– Enhance coordination in analysing reported incidents in order to pool expertise and speed up necessary corrective actions.
– Support the development of tools to ensure the traceability of medical devices and their long-term safety and performance monitoring (e.g. unique device identification systems and implant registries).
These objectives, however, are not isolated to the EU. In 2012, the Center for Devices and Radiological Health released its strategic priorities, the first of which emphasizes the complete implementation of a ‘total product lifecycle approach’. Still, the preventive and corrective maintenance of medical devices is left to be carried out by manufacturers and/or distributors and/or healthcare institutions.
3.Medical device management and maintenance: The need for evidence-based approach
As stated previously, the complexity of the medical technology assets found in healthcare is reflected in the complexity of technology management, which must be efficient so that the technology can always be used according to its intended use, safely and appropriately. The goal of medical device management and maintenance should be to achieve that every medical device is safe and accurate every time it is used on patient. To do so, a set of management and maintenance activities is being carried out. Safety and performance of medical devices is checked relative to the manufacturer’s specification, international standards and national/regional regulation. Therefore, from management perspective maintenance is a crucial aspect of the activities in a healthcare institution’s clinical engineering (CE) department because it involves significant human and financial resources. Original equipment manufacturers (OEM) recommend regular checks and tests to ensure that the safety, accuracy and precision of medical equipment are maintained throughout the product lifecycle to acceptable standards, protecting the patient [17]. This is known as maintenance. There are two main types of maintenance required for medical equipment in all hospitals: scheduled maintenance (SM) and corrective maintenance (CM). Generally, preventive maintenance can be broken down into three main categories: scheduled maintenance, performance verification and safety testing. In medical device industry, those activities are defined and performed through PMS.
Currently, management of medical devices within healthcare institutions can be considered as ‘reactive’ – responding after an event; of which there are many types ranging from complaints to those involving serious injury or in an extreme case where a serious injury or death has occurred known as ‘Vigilance’. These activities can be considered ‘passive’ as they are largely data collection activities. On the other hand, in ‘proactive’ management – endeavours meant to anticipate and curtail events before they occur; there are many types such as user surveys and manufacturer-sponsored clinical registry studies. In ‘proactive’ programme, information is actively sought to gain insight and data into the real-world performance of the device. Proactive approach can ultimately lead to predictive properties. Generally, the development and increase in the complexity of electrical equipment have led to the modernization and updating of maintenance techniques and policies. The preventive and predictive activity makes it possible to plan the shutdown, prepare the intervention team, ensure the necessary spare parts, and respectively, reduce to a minimum the parking time for repair. Maintenance planning requires the assessment of a number of parameters, including how a piece of equipment is used, how often it is used, its intended use, risk associated with its usage and its failure rates.
The maintenance of medical equipment becomes more expensive every year, and to optimize maintenance programs and reduce total cost of ownership, hospital management structures are constantly looking for solutions to extend the time of operation of equipment, in the required safety and technical performance and through the efficient use of available resources [19]. The analysis and substantiation of capital expenditures in medical organizations must be based on quantifiable factors, with a direct impact on the full associated costs. In order to optimize operating and support costs, medical technology management structures develop medical equipment maintenance programs, based on assessments and prioritization, based on risks and costs. The development of alternative plans for medical equipment takes into account the complexity and large number of existing equipment, the skills of its own specialists and their number, the technical means of calibration and control, and the budgetary resources available.
Good management of medical technologies at the hospital level can minimize malfunctions in medical devices. Proper management of medical technologies, based on increasing reliability and reducing failures, could lead to the provision of good health services in limited economic conditions. The lack of specialized technical staff, including medical bioengineers and clinical engineers in hospitals, as well as an inefficient maintenance system are the major causes that determine incidents of all kinds. This has been highlighted more than ever, during the pandemic, when hospitals and all their medical equipment were and still are overburdened [20]. During this time, more than ever, we have realized that the rules and standards of hospital care need to be improved, and maintenance policies need to be optimized as quickly as possible.
As for the data exchange, in current framework experiences gathered on the use of medical devices by users are reported to manufacturers who report certain incidents to NRAs and keep them updated on the actions taken. The NRA review the investigation undertaken by manufacturers and take further actions. This is connected to the market surveillance responsibilities of NRAs. Market surveillance comprises the total package of activities undertaken by NRAs to obtain an oversight of medical devices on the market in their territory and to ensure that the safety, quality and performance of medical devices on the market continues to be adequate. The NRA in each country may establish a mechanism for the testing of medical devices to ensure that they continue to meet their quality, safety and performance requirements. Some countries establish it under legal metrology framework.
The EU regulations establish strong and relatively detailed requirements on post-market surveillance and on how post-market surveillance data are to be used (e.g. updates to the risk management file and to clinical evaluation). The US FDA [18] has placed an increased emphasis on the possibilities for using data from experiences gained through the use of medical devices. An increasing number of regulators are considering the value of using real-world evidence for post-market surveillance, and other regulatory processes. Recent horizontal International Organization for Standardization (ISO) [4] standards for medical devices place increased emphasis on the importance of post-market surveillance. The ISO 13485 standard on quality management systems (QMS) for medical devices, used by most manufacturers, requires a post-market surveillance system to be in place. Together, these documents provide a framework for conducting post-market surveillance and using post-market surveillance data to ensure the continued quality, safety and performance of medical devices. Post-market surveillance depends upon the information i.e. the evidence that can be/is to be collected.
Safety and performance requirements of medical devices are checked relative to the environment in which medical equipment is used. This environment should be controlled in terms of temperature and humidity. There are situations in which some medical equipment is designed to be used in accordance with the specific climate of a country/region. In this case, the maintenance procedures in a particular country or region are adjusted according to these local factors. Other important factors are the age and condition of medical equipment. In case some deviation from the specification is detected preventive/corrective maintenance is being carried out. The actions required after the repair is completed include recalibration processes and a performance and safety inspection. These activities are essential for measuring device performance. Once these activities are completed, the medical equipment will be returned for use to the patient care.
In the case of corrective maintenance, a decisive role is played by the periodic technical verification, part of the preventive maintenance process. The technical file of the medical device in which any intervention made on the medical device is recorded is a reference document in corrective maintenance that helps to make decisions. Existing data in the worksheet includes the actions performed, the parts replaced, and their cost helps to identify if or when the parts need to be replaced again and helps to explain the condition of the parts during the current inspection.
In this regard, healthcare facilities need to implement evidence-based maintenance strategies, through the development of prioritization procedures aimed at a balanced assessment of relevant factors in the life of medical equipment, through an integrated approach to the elements of reliability-based maintenance, on risk-based conditions and maintenance. Although the expression “evidence-based” is well known in the medical literature, it may also be applied to maintenance. Evidence-based maintenance (EBM) [21] begins with the analysis of evidence (i.e. failures) to monitor the maintenance effectiveness and plan any necessary changes to improve it. Maintenance reports in most hospitals describe only the failures, the repair procedures and any spare parts used. What these reports never provide is information about any measures needed to prevent that failure. Evidence-based maintenance provides all the information necessary for early detection of defects, defect localization, fault diagnosis and calculation of the safe operating time of the machine.
Knowledge of the history of a failure enables the monitoring and improvement of the current maintenance strategy so that the most appropriate approach can be found. Ultimately, when the effectiveness, reliability and availability of medical equipment are improved through maintenance, the safety of staff and patients is improved. The implementation of evidence-based, or risk-based maintenance is possible for non-critical medical devices by evaluating the reliability, which can increase departmental efficiencies and streamline PM schedules. In clinical engineering departments, metrology is important to understand when testing medical devices and choosing the right biomedical test tools. Requirements for quality, safety, and competence in clinical engineering, also specifies an understanding of uncertainties, equipment implementation and traceability of measurements. Performing measurement tasks manually is much more challenging and highlights because the majority of HTM departments utilise specialist equipment. In fact, the WHO recommends utilisation of biomedical test equipment in their global medical equipment maintenance programme overview. This is because it effectively reduces clinical engineering resource, improves test accuracies and reading repeatability, when compared to traditional test and measurement methods. The hospital will also be able to provide better evidence with accurate data when evaluating and testing their alternative equipment maintenance system. This further reduces the risks and improves overall efficiencies.
4.Legal metrology in healthcare
As MD functionalities involve measurement of physiological or anatomical parameters, energy or volume of substances administered to or removed from the body, they are referred to as medical devices with measuring function (MDMF). Measurements of medical devices are obtained from the measurement devices or sensors contained within the medical device. The performance of the MDs measuring devices and sensors has a direct impact on the overall quality of the device. The quality of the device is an abstract term formalized in terms of the science of metrology seen from the definition of MDMF as follows: “MDMF is designed and produced in a way that ensures that the device provides accurate, precise and stable measurements within the limits indicated by the manufacturer and having regard to the intended purpose of the device” [22]. Discussing MDs without the direct application of metrology concepts does not make sense.
In terms of the science of metrology every measurement is just an approximation of the real value or the quantity characterized by a dimension, a unit, and a value [23]. Since it is an approximation, it means that every measurement is expressed in terms of accuracy, i.e., with its error, uncertainty and measurement traceability. The accuracy of sensors and measuring units within medical devices is tested and ensured during production. Even the best medical measuring devices and equipment are affected by wearing and tearing, environmental changes and other factors, leading to loss of accuracy over time. Every change made on the device in regular servicing activities require additional readjustments, calibrations and inspection of the entire medical device. High measurement error and uncertainty expose patients to risk or to severe hazards. Therapy-targeting changes based on inaccurate measurements cause clinical in-effectiveness to severe injuries and death outcomes. Therefore, understanding and application of metrological concepts and recognizing limitations and constraints help to interpret measurement results in healthcare whether they are used in medical diagnosis and treatment or for management of medical devices [24, 25].
The national regulators, society and every single citizen benefit from reliable and secured reference data, measurements and safety in medical diagnosis and treatment. Although regulatory control for medical devices, comprises both pre-market approval and post-market surveillance, there are significant differences between the two types of surveillance. Additionally, discrepancy is seen in a variety of methods for metrological conformity assessment testing of MDMFs cause lack of comparable dissemination of traceability. Sometimes, these metrological conformity assessment testing are commonly restricted to isolated accuracy checks of simple physical sensors. Such isolated checks ignore the complexity of the physiological measurement process and the fact that the sensor is the only one link in the measurement chain while the physical quantity being the first step towards a medically relevant measurand.
With repeated use and over a period of time, all equipment begins to degrade and that affects its accuracy and precision. In the medical device industry, a drift in the measurement is found to be unacceptable. Regularly calibrating equipment will ensure that industry defined standards are met and that the equipment is functional thus providing accurate output. Medical Equipment Calibration is carried out to minimize the uncertainty in measurements, reducing errors and bringing measurements to an acceptable level. In terms of metrology, every calibration needs to be carried out to determine device’s traceability. Traceability means that the measurement results or the deviation from a standard from the measured reference to the reported device can be traced back to guidelines based on good practice procedures. These are usually founded in national or international standards. Traceability [26] can be assured at different times and in different places. In order to maintain traceability throughout a products life cycle the necessary calibrations have to be performed by using a metrological quality standard. The concept of making a calibration traceable to a national standard can be summarised in a few key elements. The foundation of traceability in calibration is established via an unbroken chain of comparisons between its own measurements and relevant international or national standards. This link between calibrations and standards can be achieved by formally addressing the laboratories reference standards. The measurement uncertainty must be calculated for each step using appropriate means and as result the overall uncertainty of the chain can be stated. In many fields, reference materials replace physical reference standards which follow the same traceability structure as previously described. Certification of reference materials and devices are often used to demonstrate the traceability to relevant standards.
Referential measurement devices are all internal references to verify the accuracy of the measuring devices performing according to metric standards. If possible, measurement variables for the measuring instruments are defined to determine the internal reference or the standard deviation. If there is no internal reference or metric, the device is calibrated at a national measuring site or accredited laboratory. Referential measuring devices should, as a rule, be calibrated at national measurement sites or accredited calibration laboratories. This ensures that the measurement uncertainty remains within the desired limits and achieves an unbroken, documented calibration chain that ensures traceability of measurement results to national and international standards and, thus, SI units. If no measurements of a reference measurement instrument’s calibration can be traced back to a national or international measurement standard, the calibration criteria need to be documented. This also includes the documentation of natural constant or derived quantities used or acquired during the calibration.
Inspection [26] is defined as a process designed to provide objective evidence that a given item fulfils specified requirements. This is not to be mixed with calibration defined as is a procedure for detecting and fixing the uncertainties in measurements and bringing them to an acceptable level. Therefore, calibration process implies a correction being made in the device. This kind of adjustments in medical devices can only be performed by authorized distributors or services and manufacturers. On the other hand, as inspection aims to provide objective evidence that a given item fulfils specified requirements independence and impartiality of the body performing these activities are of great importance. The inspection process doesn’t imply any correction being made. If the non-compliance is detected service is required. The service is conducted by authorized distributors or services and manufacturers.
The new MDR [14, 15, 16] stresses a priority on continuous review of post-market issues and analysis of those issues, specifically that manufacturers should have “a post-market surveillance system in place which should be proportionate to the risk class and the type of device in question.” The MDR also explicitly states that data from post-market surveillance activities should feed into multiple areas, including the risk–benefit analysis, manufacturing instructions, corrective and preventive actions, and for the identification of options to improve the usability, performance and safety of the device.”
Even the new MDR recognised the gap of independence third party in post-market surveillance, still lacking standardized methods for conformity assessment testing based on metrology principles which are the only pathway towards the accomplishment of the requirements stated in the MDR. The International organization of Legal Metrology (OIML) [27] is the main institution for defining the regulatory framework for metrology characteristics and requirements of all measuring devices. Therefore, the guidelines of OIML, developed for sensors and measuring devices included in MDMF, must be considered for the development of methods for MDMF conformity assessment testing, whereas IEC 60601, IEC 62353 and ISO 80601 refer only to requirements for basic safety and essential performance the OIML guidelines address the metrological characteristics of medical devices.
Safety and performance testing of medical devices in the medical sector is one of the key factors in improving public health [28, 29, 30, 31, 32, 33, 34]. Physicians need better accurate medical measurements to diagnose diseases better, monitor patients and deliver treatments. Even an error in a simple medical device such as clinical thermometers to measure body temperature, sphygmomanometers to measure blood pressure, weighing scales to measure body weight etc. can cause medical decision making to shift from the normal decision. In some conditions, medical errors might cause patients pain bearing permanent or temporary disability and, in some states, even cause death. In another view, this matter can cause increasing health costs. Increasing the number of follow-up tests, medication and treatment for a patient who has been incorrectly diagnosed to have a certain disease will incur health costs. As a result, the accuracy of MDMFs is uncertain, their deterioration over time cannot be properly detected or prevented and the diagnostic value of their measurements becomes questionable. This significant drawback was recognized by the Institute of Electrical and Electronics Engineers (IEEE) standards group that began to work on harmonization of MD PMS performance evaluation within the Medical Device with Measuring Function (MDMF) group. The first standard the group is working on is P2727 – Standard for General Vocabulary for Conformity assessment testing of Medical Devices with Measuring Function [16]. The goal of this work is to link medical devices and their performance assessment testing to the metrological concepts and terms and to develop standards for conformity assessment testing of MDs.
5.Medical device surveillance database
As discussed, medical device pre-market and post-market requirements are very well defined globally. The existence of directives and regulations govern the overall life-cycle of medical device – from ideation to usage. Medical Device surveillance data is currently being stored in different databases such as the FDA Manufacturer and User Facility Device Experience (MAUDE) and Recall databases [35], Medical Product Safety Network (MedSun) [37] and European database on medical devices database (EUDAMED) [36]. The main purpose of the databases is to enhance traceability, cooperation and transparency regarding medical devices. Databases are designed to serve as an interaction between different stakeholders offering various functions, such as adverse event reporting program, reporting medical device problems that result in serious injury, or death and recall database. The flow of information to database, throughout the lifespan of MD, is presented in Fig. 1.
Figure 1.
Identified disadvantages and limitations of current post-market surveillance of MDs.
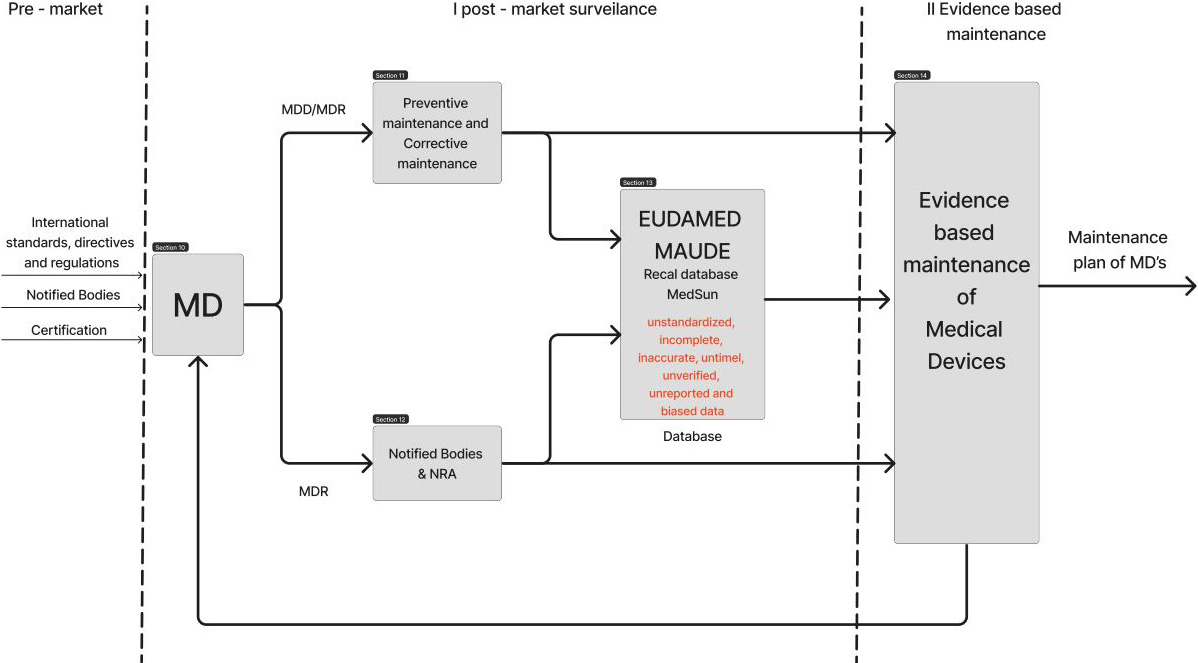
Current databases collect information for each database from both voluntary sources and mandatory reports. Judging by the number of reported incidents involving medical devices it can be concluded that the limitations of the databases are that the data submitted include “incomplete, inaccurate, untimely, unverified, or biased data.” In addition, many use errors stay underreported and data format unknown. Moreover, current databases are limited to administrative data only including manufacturers and distributor data, vigilance and clinical studies data, hence the surveillance is not taking its full capacity.
UN Sustainable Development Goal 3 states: “To ensure healthy lives and promote well-being for all at all ages.” Within this goal, a specific target is defined: “access to quality essential health-care services and access to safe, effective, quality and affordable essential medicines and vaccines for all.” In terms of above-mentioned disturbing statistics on adverse events caused by medical devices “access to safe, effective, quality and affordable medical devices” should be ensured as well. Safety and effectiveness (performance) of medical devices is indeed area in which digital transformation can deliver its full purpose.
Digitalization, which is already in place, but supported by standardized methodology for conformity assessment yielding in standard data format has the potential to solve the above identified challenge in post-market surveillance.
The introduction of standardized conformity assessment method for testing of safety and performance of medical device will produce traceable, accurate, complete, verified, nonbiased and standardized data. With this, existing digital databases will serve its purpose. Standardized conformity assessment method implies medical device quality testing by independent third-party bodies and/or manufacturers/distributors, but the evidence produces during these activities would be complete, accurate, verified, traceable and unbiased. With this approach two challenges would be solved. Firstly, safety and quality of medical devices would be inspected with calibrated etalons by independent party introducing more transparency into the overall process. Secondly, standardized data, when entered into existing databases would result in database ready for analysis using various techniques. The more analysis ready data is created the more data mining can be done. The proposed solution is presented in Fig. 2.
Figure 2.
Solution for evidence-based maintenance of medical devices during post-market surveillance.
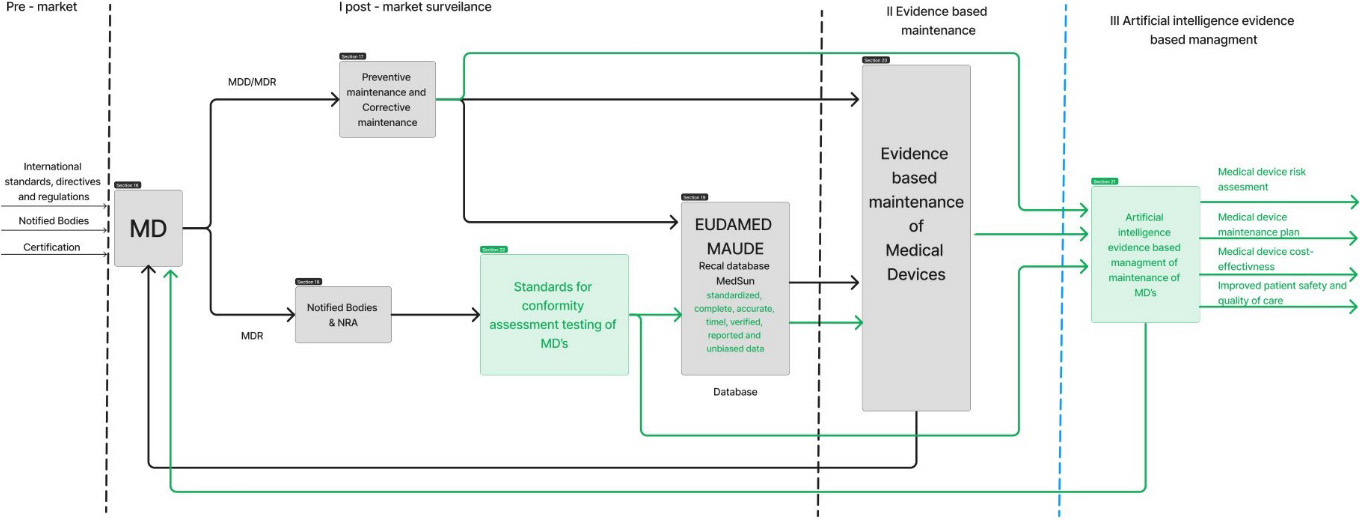
Such approach is already being validated in two countries. Evidence-based methodology for surveillance of medical devices has been part of legal metrology framework in Bosnia and Herzegovina and in Republic of Serbia. It has been introduced in the legal framework in terms of adopted bylaws. All data resulting from the inspection carried out by independent appointed inspection bodies has been entered in database specially developed for this purpose – eLab [38]. For every used medical device in healthcare institution which was subject of inspection a standardized record of safety and performance has been entered into the database periodically since inspections are conducted with determined frequency. This is step toward personalized medical device maintenance which is different than benchmarking approach as reported in some prior papers related to the evidence-based maintenance [39, 40, 41].
This practice showed that significant cost-effectiveness can be achieved in management of maintenance in healthcare institutions because standardized data showed certain patterns for healthcare institutions.
“How to understand the present and predict the future?”
Following the trend of application of artificial intelligence in other fields, as well as in healthcare, the researchers investigated application of artificial intelligence on this database as well [42, 43, 44, 45, 46, 47, 48]. The hypothesis of predictive management of maintenance of medical devices has been proved. Indeed, as medicine is shifting toward personalized medicine clinical engineering should follow the path of predictive management of medical devices.
“In God we trust, all other bring data!”
In conclusion, this article provides evidence-based examples which eliminate the identified gap. The collection of nine case reports published in this special section give an overview of the complexity of the topic and show where the successes have already been achieved, but also where the challenges of this topic lie.
References
[1] | Senbekov M et al. The recent progress and applications of digital technologies in healthcare: a review. International journal of telemedicine and applications 2020 ((2020) ). |
[2] | American College of Clinical Engineering/ACCE. Available via: https://accenet.org/about/Documents/What’s_a_Clinical_Engineer.pdf. |
[3] | World Health Organisation. Online: https://www.who.int/health-topics/medical-devices. |
[4] | International Organisation for Standardization. Online: https://www.iso.org/home.html. |
[5] | Medical device marking in China, CCC (China Compulsory Certification) [Internet]. Beijing: China Compulsory Certification; c2012 [Cited 2021 Oct 11]. Available from: http://www.ccc-cn.org/en/AboutCCC.html. |
[6] | Medical device marking in Japan, Product Safety Electrical Appliance & Material marking [Internet]. Tokyo: Japan Quality Assurance Organization; c2021 [Cited 2021 Oct 12]. Available from: https://www.jqa.jp/english/safety/service/mandatory/pse/. |
[7] | Medical device labelling obligations [Internet]. Canberra: Therapeutc Goods Administration; c2021 [Cited 2021 Oct 12]. Available from: https://www.tga.gov.au/medical-device-labelling-obligations. |
[8] | Guidance Regulating medical devices in the UK [Internet]. London: Medicines and Healthcare Products Regulatory Agency (MHRA) ; c2020 [Cited 2021 Oct 12]. Available from: https://www.gov.uk/guidance/regulating-medical-devices-in-the-uk#contents. |
[9] | Health Canada Guidance Document: Guidance for the Labelling of Medical Devices, not including in vitro diagnostic devices – Appendices for the Labelling of Soft, Decorative, Contact Lenses and Menstrual Tampons. Health Canada Guidance Document, (June 18, 2015). |
[10] | CE marking [Internet]. Brussels: European Comission; [Cited 2021 Oct 10]. Available from: https://ec.europa.eu/growth/single-market/ce-marking_hr. |
[11] | Notified bodies [Internet]. Brussels: European Commission; [Cited 2021 Oct 10]. Available from: https://ec.europa.eu/growth/single-market/goods/building-blocks/notified-bodies_en. |
[12] | The third-leading cause of death in US most doctors don’t want you to know about. https://www.cnbc.com/2018/02/22/medical-errors-third-leading-cause-of-death-in-america.html?__source=sharebar|email&par=sharebar. |
[13] | Effective post-market surveillance: Understanding and conducting vigilance and post-market clinical follow-up [Pamphlet]. London (UK): BSI Standards Ltd.; (2014) . |
[14] | EU Medical Device Regulation: Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on medical devices, amending Directive 2001/83/EC. Official Journal of the European Union, L 117, (May 05, 2017). |
[15] | EU Medical Device Regulation: Regulation (EC) No 178/2002 of the European Parliament and of the Council of 28 January 2002 laying down the general principles and requirements of food law, establishing the European Food Safety Authority and laying down procedures in matters of food safety. Official Journal of the European Communities, L 31, (February 01, 2002). |
[16] | Badnjević A. et al. Post-market Surveillance of Medical Devices: A Revie. Technology and Health Care. (2022) ; 30: (6): 1315-1329. |
[17] | Iadanza E, Gonnelli V, Satta F. et al. Evidence-based medical equipment management: a convenient implementation. Med Biol Eng Comput. (2019) ; 57: : 2215-2230. doi: 10.1007/s11517-019-02021-x. |
[18] | Food and Drug Administration (FDA) [Internet]. Silver Spring (MA): Food and Drug Administration; [Cited 2021 Oct 11]. Available from: https://www.fda.gov/. |
[19] | Corciovǎ C, Fuior R, Andriţoi D, Luca C. Assessment of Medical Equipment Maintenance Management. In: Márquez FPG, editor. Operations Management [Working Title] [Internet]. London: IntechOpen; 2022 [cited 2022 Nov 06]. Available from: https://www.intechopen.com/online-first/1085559. doi: 10.5992/intechopen.1000210. |
[20] | Badnjeviá A, Pokvić LG, Džemić Z. et al. Risks of emergency use authorizations for medical products during outbreak situations: a COVID-19 case study. BioMed Eng OnLine. (2020) ; 19: : 75. doi: 10.1186/s12938-020-00820-0. |
[21] | Wang B, Fedele J, Pridgen B, Williams A, Rui T, Barnett L, Granade C, Helfrich R, Stephenson B, Lesueur D, Huffman T, Wakefield J, Hertzler L, Poplin B. Evidence-Based Maintenance: Part I: Measuring Maintenance Effectiveness With Failure Codes. Journal of Clinical Engineering. (2010) ; 35: : 132-144. doi: 10.1097/JCE.0b013e3181e6231e. |
[22] | Medical Device with Measuring Function. Online: https://ec.europa.eu/docsroom/documents/10283/attachments/1/translations/en/renditions/native. |
[23] | Brown RJ. Measuring measurement-What is metrology and why does it matter? Measurement. (2021) ; 168: . doi: 10.1016/j.measurement.2020.108408. |
[24] | Monteiro EC, Leon LF. Metrological reliability of medical devices. Journal of Physics: Conference Series. (2015) ; 588: : 20-32. |
[25] | Badnjević A, Cifrek M, Magjarević R, Džemić Z. (eds) Inspection of Medical Devices. Series in Biomedical Engineering. (2018) ; Springer, Singapore. |
[26] | International Vocabulary of Metrology – Basic and General Concepts and Associated Terms (VIM) – https://www.oiml.org/en/files/pdf_v/v002-200-e07.pdf. |
[27] | Organisation Internationale de Métrologie Légale (OIML). 2015. National metrology systems – Developing the institutional and legislative framework. Accessed December 15, 2021. https://www.oiml.org/en/files/pdf_d/d001-e20.pdf. |
[28] | Badnjevic A, Gurbeta L, Jimenez ER, Iadanza E. Testing of mechanical ventilators and infant incubators in healthcare institutions. Technology and Health Care. (2017) ; 25: (2), 237-250. |
[29] | Gurbeta L, Dzemic Z, Bego T, Sejdic E, Badnjevic A. Testing of Anesthesia Machines and Defibrillators in Healthcare Institutions. J Med Syst. (2017) ; 41: : 133. doi: 10.1007/s10916-017-0783-7. |
[30] | Gurbeta L, Badnjevic A, Dzemic Z, Jimenez E.R, Jakupovic A. Testing of therapeutic ultrasound in healthcare institutions in Bosnia and Herzegovina, 2nd EAI International Conference on Future Access Enablers of Ubiquitous and Intelligent Infrastructures, 24-25 October 2016, Belgrade, Serbia. |
[31] | Gurbeta L., Alic B. Dzemic Z, Badnjevic A. ((2017) ) Testing of infusion pumps in healthcare institutions in Bosnia and Herzegovina. In: Eskola H, Väisänen O, Viik J, Hyttinen J. (eds) EMBEC & NBC 2017. EMBEC 2017, NBC 2017. IFMBE Proceedings, vol 65. Springer, Singapore. |
[32] | Gurbeta L, Alic B, Dzemic Z, Badnjevic A. ((2017) ) Testing of dialysis machines in healthcare institutions in Bosnia and Herzegovina. In: Eskola H, Väisänen O, Viik J, Hyttinen J. (eds) EMBEC & NBC 2017. EMBEC 2017, NBC 2017. IFMBE Proceedings, vol 65. Springer, Singapore. |
[33] | Gurbeta L, Vukovic D, Dzemic Z, Badnjevic A. Legal metrology procedures for increasing safety and performance characteristics with cost benefits analysis: Case study dialysis machines, IUPESM – The World Congress on Medical Physics & Biomedical Engineering in Prague, June 3–8, 2018. |
[34] | Gurbeta L, Dzemic Z, Badnjevic A. Establishing traceability chain of infusion and perfusor pumps using legal metrology procedures in Bosnia and Herzegovina, IUPESM – The World Congress on Medical Physics & Biomedical Engineering in Prague, June 3–8, 2018. |
[35] | Kavanagh KT, Brown RE, Jr., Kraman SS, Calderon LE, Kavanagh SP. Reporter’s occupation and source of adverse device event reports contained in the FDA’s MAUDE database. Patient Related Outcome Measures. (2019) ; 10: : 205. |
[36] | EUDAMED Database https://ec.europa.eu/tools/eudamed/eudamed. |
[37] | MEDSun https://www.fda.gov/medical-devices/medical-device-safety/medsun-medical-product-safety-network. |
[38] | Gurbeta L, Badnjević A. Inspection process of medical devices in healthcare institutions: software solution. Health Technol. (2017) ; 7: (1): 109-117, doi: 10.1007/s12553-016-0154-2. |
[39] | Li Y et al., Development of a conceptual benchmarking framework for healthcare facilities management: Case study of Shanghai municipal hospitals. Journal of Construction Engineering and Management. (2020) ; 146: (1): 05019016. |
[40] | Neslišah AKAR, Yekta Ü, Esin Ö. A Comprehensive Medical Equipment Management Software System for Increased Patient Safety. IEEE International Symposium on Medical Measurements and Applications (MeMeA). IEEE, (2019) . |
[41] | Corciovǎ C, Doru A, Cǎtǎlina L. A modern approach for maintenance prioritization of medical equipment. Oper Manag-Emerg Trend Digit Era, (2020) . |
[42] | Almir B, Lejla GP, Mehrija H, Lejla B, Zerina M, Živorad K, Jasmin K, Leandro P. Evidence-based clinical engineering: Machine learning algorithms for prediction of defibrillator performance, Biomedical Signal Processing and Control. September (2019) ; 54: : 101629. |
[43] | Kovačević Ž, Gurbeta Pokvić L, Spahić L, Badnjevic A. Prediction of medical device performance using machine learning techniques: infant incubator case study. Health Technol. (2019) ; doi: 10.1007/s12553-019-00386-5. |
[44] | Badnjević A, Avdihodžić HI, Gurbeta P. Artificial intelligence inmedical devices: past, present and future. Psychiatria Danubina. (2021) ; 33: (suppl 3): 101-106. |
[45] | Spahić L, Kurta E, Cordic S, Becirovic M, Gurbeta L, Kovacevic Z, Izetbegovic S, Badnjevic A. Machine Learning Techniques for Performance Prediction of Medical Devices: Infant Incubators. In: Badnjevic A, Škrbić R, Gurbeta Pokvić L. (eds) CMBEBIH 2019. CMBEBIH 2019. IFMBE Proceedings, vol 73. Springer, Cham, 2020. |
[46] | Hrvat F, Spahic L, Gurbeta Pokvic L, Badnjevic A. Artificial Neural Networks for prediction of medical device performance based on conformity assessment data: Infusion and perfusor pumps case study, IEEE 9th Mediterranean Conference on Embedded Computing (MECO), 08–11 June 2020, Budva, Montenegro. |
[47] | Deumić A, Imamović E, Pokvić LG, Badnjević A. Performance Inspection of Patient Monitors According to the Legal Metrology Framework: Bosnia and Herzegovina Case Study. In: Badnjevic A, Gurbeta Pokvić L (eds) CMBEBIH 2021. CMBEBIH 2021. IFMBE Proceedings, vol 84. Springer, Cham. doi: 10.1007/978-3-030-73909-6_44. |
[48] | Gurbeta Pokvic L, Deumic A, Lutovac B, Badnjevic A. Possibility of Managing Medical device Post-market Surveillance using Artificial Intelligence and Standardized Methodology, IEEE 10th Mediterranean Conference on Embedded Computing (MECO), 07–10 June 2021, Budva, Montenegro. |

